What happens when a mechanism meant to protect the cell gets exploited by bad-mannered viruses?
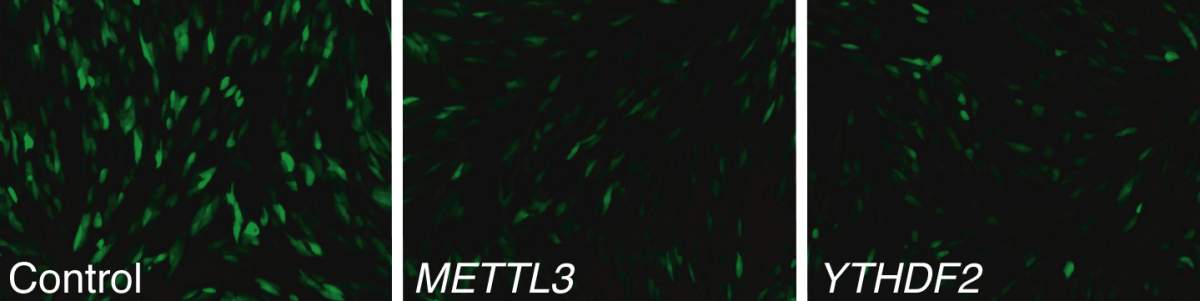
Human cells infected with a CMV virus (green), in a control sample and in cells missing enzymes belonging to the m6A system (center and right)
Viruses can be ill-mannered guests, freely helping themselves to the host cells’ machinery and using it to make more viruses. Weizmann Institute of Science researchers went looking for a particular event that was thought to improve the viruses’ abilities to infect cells; they found it occurring in a surprising place, suggesting that an entirely different mechanism – one actually meant to protect cells – can aid and abet this invasion. The group’s findings, which were reported in Nature Immunology, provide new insights into the intricate relations between host cell and virus, and possibly to new research on ways of treating infection or mistaken immune reactions.
The event the group was seeking is a modification to a strand of RNA – instructions copied from the genome. Scientists discovered that RNA can undergo modification after it is produced in the cell nucleus and before it reaches its goal in the cell. The function of that RNA – whether it has been made by a virus that has inserted its DNA into a cell nucleus or the genes belonging to the cell – will be changed as a result. The modification Dr. Noam Stern-Ginossar and her lab group in the Institute’s Molecular Genetics Department investigated is one of the more common RNA modifications – m6A – which occurs in something like a quarter of all RNA, viral and human alike. This modification involves an enzyme that attaches a compound known as a methyl group (the “m”) to a particular site on the RNA (the 6A).

(l-r) Roni Winkler, Julie Tai-Schmiedel, Aharon Nachshon and Dr. Noam Stern-Ginossar
Research on this modification had suggested that many viruses use it to infect cells, and the Institute scientists had therefore expected to find the enzyme attaching methyl groups to the viral RNA. They thus began their investigation from the viruses’ point of view. They inserted CMV, a giant virus that inserts its DNA into nuclei, into human cells grown in the lab. When they deleted the enzyme that is known to generate the m6A modification, the virus did not replicate and spread. This suggested that m6A does, indeed, play a role in the infection process.
Next, working with departmental colleague Dr. Schraga Schwartz and his group, the researchers created two lines of cells ‒ with and without the ability to modify RNA ‒ infected them with the virus and mapped the sites on the viral RNA that were open to the m6A modification, identifying several. But when they compared the viral RNA from the mutated cells – those without the ability to modify the RNA – to that of the normal ones, they were surprised to find little difference between the two. In contrast, there was a large difference between the RNA of the two lines of host RNA themselves: The mutated cells showed a much stronger expression of genes with anti-viral activity.
This was puzzling: Why would the normal cells have a mechanism that enabled the viruses to multiply and spread? With further investigation, the team discovered that these modifications mainly occurred in the RNA produced from a single gene – the one for the antiviral substance, interferon.
There is a continual trade-off between killing the virus and protecting the host
Interferon is the body’s first line of defense against infection. It is a powerful anti-viral, but it is a very broad killing machine. “Every cell in our bodies has sensors that can detect a virus and release interferon in response,” says Stern-Ginossar. “This is part of the innate immune system, and it affects both the infected cell and its neighbors, sounding an alarm and holding off the infection as best it can until the adaptive immune system can identify the virus and learn how to fight it.”
After further experimentation, the research group came up with a possible explanation for the improved success of the virus in cells with normally occurring m6A: The modification turns down the production of interferon. “Interferon, like most of the substances produced by the immune system, is toxic. Too much can kill the cell,” explains Stern-Ginossar. “So there is a continual trade-off between killing the virus and protecting the host. Because the amount is so crucial, this substance has a very fast ‘on-off’ switch. We think m6A acts as the ‘off’ switch. When this modification did not occur, interferon was produced continuously and the virus was almost completely inhibited.”
The group then experimented with additional cell-and-virus combinations, finding that all the different viruses they tested triggered interferon production, and they reproduced only when m6A turned down production of this strong anti-viral substance. In collaboration with Prof. Jacob Hanna of their department, the team then created genetically engineered mice in which the m6A enzyme was mutated, and here too, they found the same phenomenon they had seen in the cell cultures: When m6A did not occur, the levels of interferon increased to dangerous levels in the mice, but the viral infection was held back.
“We think that much of the research reporting m6A as a modification to the viral RNA for the purpose of propagation may really have been observing the viruses taking advantage of a protective mechanism in the host cells – one that leaves the surviving viruses a window of opportunity in which to multiply,” says Stern-Ginossar. “Our findings create a general picture of the initial stages of viral infection that could lead to new ideas about treating or preventing viral illnesses.” In addition, they might provide insight into certain autoimmune diseases that are tied to mistakes in the RNA, inflammation and the mechanisms for regulating substances like interferon, over which the body must exercise tight control.
Dr. Noam Stern-Ginossar’s research is supported by the Sir Charles Clore Research Prize; the American Committee for the Weizmann Institute of Science 70th Anniversary Lab; Nona L. Abrams; Ms. Marla Bergman; and the Alan and Laraine Fischer Foundation. Dr. Stern-Ginossar is the incumbent of the Skirball Chair in New Scientists.
Dr Schraga Schwartz’s research is supported by the David and Fela Shapell Family Foundation INCPM Fund for Preclinical Studies; the Kahn Family Research Center for Systems Biology of the Human Cell; the estate of Emile Mimran; and the European Research Council. Dr. Schwartz is the incumbent of the Robert Edward and Roselyn Rich Manson Career Development Chair in Perpetuity.
Prof. Jacob Hanna’s research is supported by the Nella and Leon Benoziyo Center for Neurological Diseases; the David and Fela Shapell Family Center for Genetic Disorders Research; the Kekst Family Institute for Medical Genetics; the Helen and Martin Kimmel Institute for Stem Cell Research; the Helen and Martin Kimmel Award for Innovative Investigation; Pascal and Ilana Mantoux; the Edmond de Rothschild Foundations; the Zantker Charitable Foundation; Rachel Peles; the estate of Zvia Zeroni; and the European Research Council.
Recent Comments